
In this episode of the VitaDAO Aging Science Podcast I spoke with Dr. Dan Ehninger about the shortcomings of modern mouse research as he sees them. I share many of the same concerns and think we do have to ensure that the foundations of biogerontology are sound. Although I do believe that the time for clinical translation is now, in parallel we must work on these foundational questions, some of which include:
- Does rapamycin and caloric restriction really work and are these treatments robust? And what do we mean when we say “really” and “robust”?
- The mouse is a fundamentally flawed model due to its shortlifespan and lab adaptations – but how flawed is it? Do the flaws preclude meaningful conclusions in aging research?
- How to design mouse studies that mitigate these flaws?
- How do we measure aging, functional decline, aging rate outside of lifespan? (e.g. using age-sensitive phenotypes; ASPs, to use a term from Dan’s research)
Although I am a longevity optimist and may not always agree with all the points that Dan makes, I think it is important for skeptics to be heard. It was a pure coincidence that I stumbled on his work, which some of my colleagues have never heard of. After reading several of his papers, I decided it is time to highlight his outstanding and controversial work as a service to the community.
Dr. Dan Ehninger – Brief Bio
Dan Ehninger is a Senior Research Group Leader at the German Center for Neurodegenerative Diseases (DZNE) in Bonn. His group is interested in the biology of aging and the pathogenesis of age-related brain disorders. He studied medicine at Charité University Medicine/Berlin, Harvard Medical School/Boston and University College London. From 2001 to 2004, he carried out his graduate work in the laboratory of Gerd Kempermann at the Max Delbrück Center for Molecular Medicine in Berlin, where he worked on the behavioral regulation of adult hippocampal neurogenesis and adult cell formation in the neocortex. His postdoctoral work (2004 to 2009) with Alcino J. Silva at University of California - Los Angeles focused on molecular and cellular mechanisms of cognitive impairments in mouse models of neuropsychiatric disorders. In 2010, Dan Ehninger joined the faculty of the German Center for Neurodegenerative Diseases (DZNE) and was later promoted to a tenured Senior Research Group Leader position.

https://www.dzne.de/en/research/research-areas/fundamental-research/research-groups/ehninger/research-areasfocus/
Mice are very short-lived
There are three major issues with mice as a model:
1) They are shorter-lived than expected based on their weight – in the wild early death is compensated by fast reproduction. This means the “tricks” we use to extend mouse lifespan might not work on humans, because we already evolved to utilize these enhancements and more. One solution is to learn from longer-lived animals, which is what comparative biogerontologists do. We discussed this topic in an earlier podcast with Vera Gorbunova.
2) Mice develop cancer more readily than humans (probably related to point 1), which is the key reason why Dan thinks they fail to faithfully capture aspects of human aging. At least if all you do is measure lifespan.
3) Strains we use in research labs are further adapted to fast growth and reproduction. Thus reproducing and growing even faster than in the wild. One solution to this problem is using more diverse strains, as discussed with Rich Miller in an earlier podcast.
Why do we still run mouse studies? Because using mice is better than the alternatives like cell culture, invertebrates, reading tea leaves, etc. Whereas the truly superior alternatives are not feasible or affordable. Did you know that a large phase III study in humans targeting mortality, as is often done for cardiovascular drugs and will be necessary for anti-aging treatments, can cost north of 100 million dollars?
Healthspan and lifespan uncoupling
There are two schools of thought on the matter of healthspan. Some people believe that it is difficult or impossible to extend lifespan substantially without improving health. Others believe that by now we have so many examples of healthspan-lifespan uncoupling that we must consider these almost independent.
To give an example of such healthspan-lifespan uncoupling in mice we do not need to look very far. A recent study found that reducing the levels of the famous Myc oncogene in mice extends lifespan by reducing cancer while leading to premature aging-like phenotypes (Wang et al. 2023). These include fatty liver, reduced strength, fur quality and spinal degeneration.
To give some more real-world examples of healthspan-lifespan uncoupling in humans, women live longer than men while having more age-related diseases. Testosterone replacement therapy could improve quality of life in older men even though it worsens CVD risk factors, but the effects on mortality are controversial (1).
No matter which school you belong to, there is emerging consensus that longevity treatments need to improve health and affect reasonable biomarkers of aging, especially functional ones.
The rate and trajectory of change in these markers can teach you a lot about the underlying mechanism of your intervention. That discussion is at the heart of our podcast episode.
Disease modifying drugs vs. Band-Aid solutions
I like to use these terms from pharmacology to think about potential longevity drugs and treatments.
“Disease modifying drugs” are pharmacological agents that aim to modify the underlying pathophysiology of a disease, potentially slowing down or halting the progression of the disease. In contrast, non-disease modifying drugs typically focus on symptom management without significantly influencing the underlying disease progression. For example, in the case of Alzheimer’s disease a cholinesterase inhibitor such as donepezil would be symptomatic, whereas anti amyloid antibodies should be disease modifying. For osteoarthritis painkillers are typical examples of a symptomatic solution whereas senolytics – it is hoped – could be disease modifying.
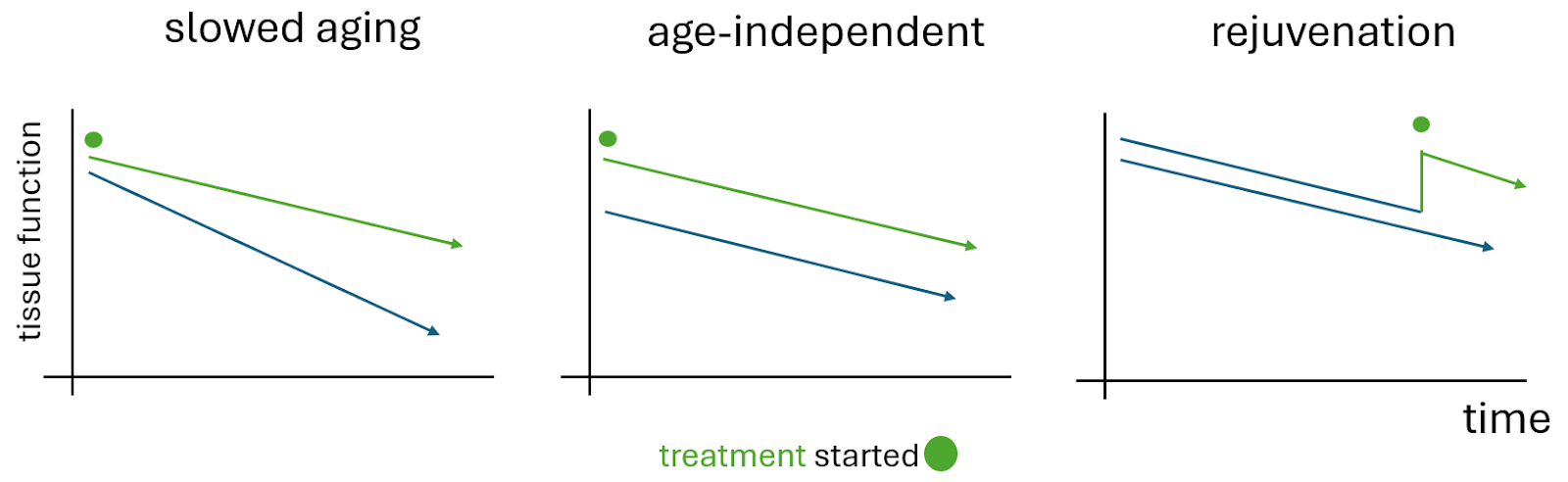
Dan’s argument, as I understand it, is that several lifespan extending treatments have age-independent effects on aging-related phenotypes. He calls them age-sensitive phenotypes; ASPs. This was something his group saw for mTOR hypomorphism, dwarfism and intermittent fasting in mice (Xie et al. 2022). Based on this age-independent effect he speculates that these treatments do not slow aging and instead extend lifespan by delaying cancer. All perfectly reasonable, although I do have some questions even after the podcast.
Why would these treatments improve ASPs in young animals? I would presume if they did nothing for aging the null hypothesis would be that some ASPs go down and others go up. Maybe we can discuss this the next time! Another issue that I find counterintuitive is that usually such “band-aid” solutions do NOT alter the course of a disease and the benefits do NOT persist over a long time and well into old-age. Normally, I would expect a convergence towards the expected path with a non-disease modifying drug or treatment. This is what I would call a “band-aid solution” but rapamycin and CR seem to be much more than that.
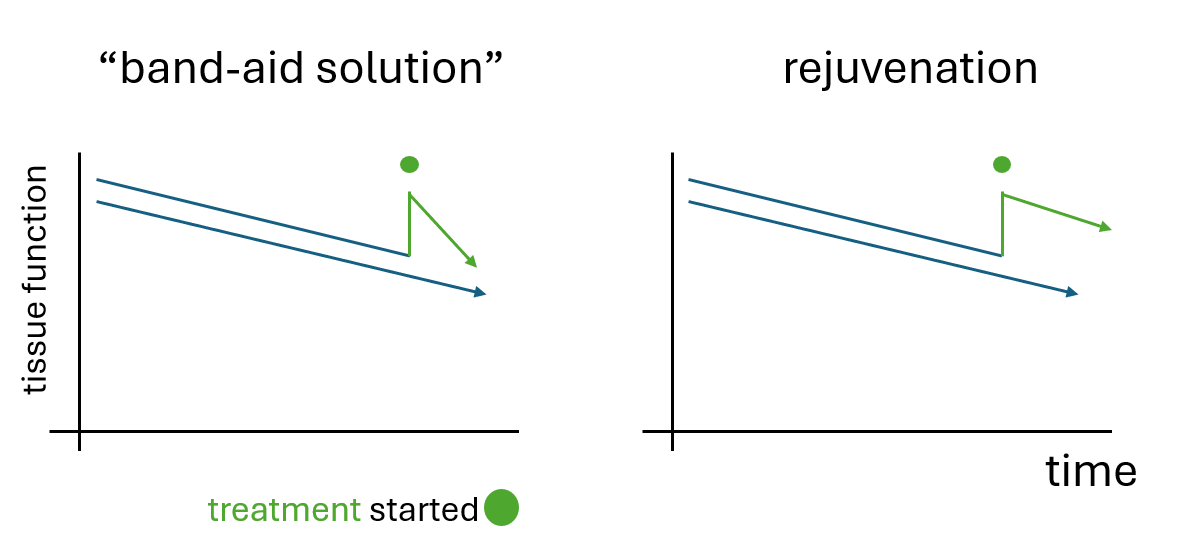
Many would argue that the actual mechanism of action does not matter. “If it works it works”. Caution is warranted here, however. We only know that these treatments work in mice and mouse cancer incidence is much higher than in humans (2). It also matters in the long term. When developing robust therapies we need to build on solid foundations. If CR only operated through cancer inhibition than all the effort to search for CR-mimetic drugs in order to slow aging would be wasted.
Wider context and reception
While the points made by Dan might seem controversial at first glance they are being echoed by many other researchers. Not everyone agrees on the specifics, but there is emerging consensus that we have problems with our model systems and need to improve. Steve Austad, for example, is a big proponent of studying the longevity mechanisms employed by ultra long-lived species rather than short-lived mice. We will interview him soon on this topic. Similarly, Peter Fedichev believes that the mouse is not an adequate model and he focuses on data mining human datasets instead to find new gerotherapeutics. Rich Miller believes that currently used mouse strains lack genetic diversity and we should focus on using heterogeneous strains while Arlan Richardson would like to validate treatments in rats, that are even more genetically distinct from regular lab mice.
Summary and conclusions
Ultimately, I do think that the high incidence of cancer in mice exaggerates the benefits of certain treatments, in agreement with Dan. There are, however, two interesting counter arguments to Dan’s theory which states that almost all the benefits of longevity interventions are due to cancer. 1/ If it was so easy than many more drugs should extend lifespan. Plenty of drugs tested for lifespan extension have strong chemoprevention data, either theoretical or preclinical, yet they fail to extend lifespan. The only drug that works robustly is coincidentally in a major growth sensing pathway, mTOR, which is also decreased by several other canonical interventions like dwarfism and calorie restriction. 2/ Taueber’s paradox dictates that the wholescale eradication of cancer in humans would only lead to a 5% increase in lifespan because other age-related diseases progress at the same time. In a similar vein, the attenuation of cancer in mice might not be enough for substantial lifespan extension, although this would require modelling studies to prove conclusively.
I also agree with Dan that our mouse studies would be much better if we included young treated controls too. Researchers with sufficient funding should run such controls much more often.
I think – and that is my personal speculation – that rapamycin is an odd drug that leads to partial rejuvenation and does not operate by slowing aging per se. This would explain some of the results seen in Dan’s data (see Xie et al. 2022 for mTOR hypomorphic mice, see Neff et al. 2013 for rapamycin).
Indeed, it seems that there is a causal pathway between fast growth and fast aging. The pathways that enable fast growth, tissue regeneration and reproduction may be harmful in the long run. There is a switch-like aspect to CR and CR-like states, which is consistent with the idea they may have evolved as a response to acute nutrient starvation and famines. Perhaps this can explain why they are beneficial without slowing all aspects of aging?
This and many other questions remain to be answered. Let’s get on with the work so we can slow aging as effectively as possible in humans.
Further reading
(1) It is possible that high dose testosterone therapy is beneficial while regular HRT is actually beneficial: “Fewer deaths occurred with testosterone treatment (six [0·4%] of 1621) than placebo (12 [0·8%] of 1537) without significant differences between groups (odds ratio [OR] 0·46 [95% CI 0·17–1·24]; p=0·13).”
https://www.thelancet.com/journals/lanhl/article/PIIS2666-7568(22)00096-4/fulltext
(2) Cancer could bias almost every single functional outcome and aging phenotype except perhaps histopathology. Imagine a mouse with underlying cancer. Will it have high or low frailty? Will it cooperate in a water maze or have high grip strength?
Our manuscript with Prof. Brian Kennedy and Matt Kaeberlein on the importance of long-lived mouse controls which would help to mitigate some of the “artefactual” or “idiosyncratic” non-age related deaths in mice:
Pabis, Kamil Konrad, et al. "The impact of short-lived controls on the interpretation of lifespan experiments and progress in geroscience." bioRxiv (2023): 2023-10.
https://www.biorxiv.org/content/10.1101/2023.10.08.561459v1.abstract
CR Plus, CR adjacent, CR independent - what the heck?
A blog post in which I worry about the lack of new models in mouse aging since most of them seem to be anti-anabolic and CR-adjacent.
https://biogerontolgy.blogspot.com/2020/09/cr-plus-cr-adjacent-cr-independent-what.html?q=anabolism
Keshavarz, Maryam, et al. "Targeting the “hallmarks of aging” to slow aging and treat age-related disease: fact or fiction?." Molecular Psychiatry 28.1 (2023): 242-255.
Note: a recent review where Dan goes into his arguments
Xie, Kan, et al. "Deep phenotyping and lifetime trajectories reveal limited effects of longevity regulators on the aging process in C57BL/6J mice." Nature Communications 13.1 (2022): 6830.
Keshavarz, Maryam, et al. "Targeting the “hallmarks of aging” to slow aging and treat age-related disease: fact or fiction?." Molecular Psychiatry 28.1 (2023): 242-255.
Keyfitz, Nathan. "What difference would it make if cancer were eradicated? An examination of the Taeuber paradox." Demography 14 (1977): 411-418.
https://link.springer.com/article/10.2307/2060587
Neff, Frauke, et al. "Rapamycin extends murine lifespan but has limited effects on aging." The Journal of clinical investigation 123.8 (2013): 3272-3291.
Ham, Daniel J., et al. "Distinct and additive effects of calorie restriction and rapamycin in aging skeletal muscle." Nature communications 13.1 (2022): 2025.
Wang, Huabo, et al. "Premature aging and reduced cancer incidence associated with near-complete body-wide Myc inactivation." Cell reports 42.8 (2023).


